欧代注册无需技术文件?模板与工厂自行处理的风险解析
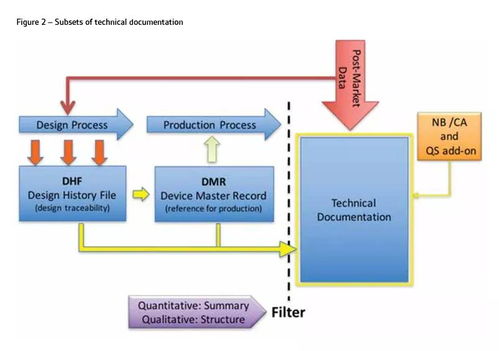
随着欧盟市场监管法规的强化,欧洲授权代表(欧代)注册成为出口欧盟产品的必备环节。一些企业可能听信‘欧代注册无需技术文件,只需套用模板由工厂自行搞定’的说法,但这实际上存在重大合规隐患。从法律层面看,欧盟法规如医疗器械法规(MDR)、体外诊断器械法规(IVDR)及通用产品安全指令(GPSD)均明确要求,欧代注册必须基于完整的技术文件,包括产品规格、测试报告、风险评估和符合性声明等。这些文件是证明产品符合欧盟安全标准的核心证据,若缺失或仅靠模板填充,可能导致注册无效,甚至面临产品下架、罚款或法律诉讼。
依赖工厂自行处理模板的做法虽看似便捷,但风险极高。工厂往往缺乏对欧盟法规的深度理解,可能忽略技术细节或更新要求,导致文件与产品实际不符。例如,医疗器械的分类、临床评估或软件验证等复杂内容,模板难以覆盖所有场景,易引发监管审查失败。更严重的是,如果产品因文件问题造成安全事故,企业需承担全部责任,欧代也可能被追责。
因此,企业应选择专业合规服务,确保技术文件真实、完整且定期更新。切勿为省事而冒险,合规才是长远发展的基石。
如若转载,请注明出处:http://www.supprt-apple.com/product/49.html
更新时间:2026-02-28 14:32:08









